 Людям із порушенням зору
Людям із порушенням зору  In English
In English Маркування медичних виробів згідно вимог законодавства
Опубліковано 15.06.2021 о 12:21Державна служба з лікарських засобів та контролю за наркотиками у Львівській області інформує про вимоги законодавства щодо маркування медичних виробів.
Маркування медичних виробів затверджується технічними регламентами:
- «Технічний регламент щодо медичних виробів» затверджений Постановою КМУ від 2 жовтня 2013р. № 753.
- «Технічний регламент щодо медичних виробів для діагностики in vitro» затверджений Постановою КМУ від 2 жовтня 2013р. № 754.
- «Технічний регламент щодо активних медичних виробів, які імплантують» затверджений Постановою КМУ від 2 жовтня 2013р. № 755.
Також маркування медичних виробів повинно відповідати вимогам Законів України:
- Закон України “Про захист прав споживачів”;
- Закон України “Про загальну безпечність нехарчової продукції”;
- Закон України “Про забезпечення функціонування української мови як державної”
Щодо маркування медичних виробів діють Національні стандарти України:
- ДСТУ EN 980:2007 “Символи графічні для маркування медичних виробів”;
- ДСТУ 3798-98 (IEC 60601-1:1988) Вироби медичні електричні. Частина 1. Загальні вимоги безпеки (для активних виробів).
Маркуватися медичні вироби, які знаходяться в обігу на території України, повинні на державній мові (згідно вимог ЗУ «Про забезпечення функціонування української мови як державної»).
Загальні вимоги до маркування медичних виробів регламентовані пунктами 42-47 Розділу ІІ додатку І постанови КМУ від 2 жовтня 2013 р. № 753 «Про затвердження Технічного регламенту щодо медичних виробів», а саме:
Кожний медичний виріб повинен супроводжуватися інформацією, необхідною для його безпечного та правильного застосування з урахуванням рівня підготовки та кваліфікації споживачів і користувачів, а також для ідентифікації виробника.
Зазначена інформація включає в себе інформацію на етикетці та інструкцію із застосування.
Наскільки це можливо і доцільно інформація, необхідна для безпечного застосування медичного виробу, розміщується безпосередньо на медичному виробі та/або на упаковці кожної одиниці медичного виробу чи в разі потреби – на упаковці для продажу. Якщо індивідуальне пакування кожної одиниці неможливе, то ця інформація розміщується на вкладці, яка додається до одного або більше медичних виробів.
Інструкція із застосування вкладається в упаковку кожного медичного виробу. Такі інструкції із застосування не є необхідними для медичних виробів, що відносяться до класу I або IIа, якщо ці медичні вироби можуть безпечно використовуватися без такої інструкції.
У разі можливості інформація, що має супроводжувати медичні вироби, повинна наводитися у формі символів. Усі використані символи та ідентифікаційні кольори повинні відповідати гармонізованим стандартам (прим.: нижче будуть розібранні найпоширеніші символи). У разі відсутності таких стандартів використані символи та кольори повинні бути описані в документації, що постачається разом з медичними виробами.
Етикетка медичного виробу повинна містити такі елементи:
1) найменування або торгову марку і місцезнаходження виробника. Для медичних виробів, що імпортуються з метою введення в обіг, на етикетці, або зовнішньому пакуванні, або в інструкції із застосування додатково зазначається найменування та місцезнаходження уповноваженого представника, якщо виробник не є резидентом України;
2) дані, що обов’язково необхідні для ідентифікації медичного виробу, а також вміст пакування;
3) в разі потреби – слово “Стерильно”;
4) в разі потреби – код партії (після слова “Партія”) чи серійний номер;
5) в разі потреби – строк, до якого гарантовано безпечне застосування медичного виробу, із зазначенням року та місяця;
6) в разі потреби – позначку про призначення для одноразового застосування. Позначка виробника про одноразове застосування медичного виробу має відповідати гармонізованим стандартам;
7) для медичних виробів, виготовлених на замовлення, – слова “медичний виріб виготовлено на замовлення”;
8) для медичних виробів, призначених для клінічних досліджень, – слова “виключно для клінічних досліджень”;
9) інформацію про будь-які спеціальні умови зберігання медичного виробу та/або використання;
10) інформацію про будь-які спеціальні інструкції з експлуатації;
11) інформацію про будь-які запобіжні заходи та/або застереження;
12) рік виготовлення – для активних медичних виробів, на які не поширюється дія підпункту 5 цього пункту. Така інформація може бути включена в номер партії або серійний номер;
13) в разі потреби – інформацію про метод стерилізації;
14) якщо медичний виріб містить як невід’ємну частину похідні крові людини – відповідне позначення, що медичний виріб містить похідні крові людини.
Якщо цільове призначення медичного виробу не є очевидним для користувача, виробник повинен чітко зазначити його на маркуванні та в інструкції із застосування.
У разі доцільності і можливості медичні вироби та їх компоненти, що знімаються, мають бути ідентифіковані (у разі потреби – за номерами партій) для забезпечення виявлення будь-якого потенційного ризику, пов’язаного з такими медичними виробами та їх компонентами, що знімаються.
У разі потреби в інструкції із застосування зазначаються:
1) відомості, зазначені в пункті 44 цього розділу (крім підпунктів 4 і 5) цього додатка;
2) експлуатаційні властивості, зазначені в пункті 3 розділу I цього додатка, та будь-які небажані побічні дії;
3) якщо медичний виріб для використання за призначенням має бути встановлений або з’єднаний з іншими медичними виробами та/або обладнанням – детальний опис характеристик цього медичного виробу, достатній для правильного підбору таких медичних виробів та/або обладнання для безпечного спільного застосування;
4) повний обсяг інформації, необхідної для перевірки правильності встановлення медичного виробу і можливості його правильного і безпечного використання, разом з відомостями про характер та періодичність технічного обслуговування і калібрування для забезпечення його точної і безпечної роботи протягом всього строку експлуатації;
5) у разі потреби – інформація, необхідна для уникнення певних ризиків, пов’язаних з імплантацією медичного виробу;
6) інформація щодо ризиків виникнення взаємних перешкод, пов’язаних з присутністю медичного виробу під час проведення спеціальних досліджень або лікування;
7) необхідні інструкції на випадок пошкодження стерильного пакування та в разі потреби – інформація щодо методів рестерилізації;
8) якщо медичний виріб може бути повторно використаний – інформація щодо відповідних процесів підготовки до повторного застосування, зокрема щодо очищення, дезінфекції, пакування, способу стерилізації для медичного виробу, що підлягає рестерилізації, та будь-яких обмежень кількості повторних застосувань.
Якщо медичні вироби потребують стерилізації перед застосуванням, інструкції з очищення та стерилізації мають бути такими, щоб за умови правильного виконання зазначені вироби продовжували відповідати вимогам розділу I цього додатка.
Якщо медичний виріб призначено для одноразового використання, зазначається інформація про відомі характеристики та технічні фактори, які відомі виробникові і які можуть становити ризик за умови повторного використання медичного виробу. Якщо відповідно до пункту 42 розділу II цього додатка інструкції з використання не потрібні, інформація має надаватися на запит користувача;
9) докладна інформація щодо будь-якої додаткової обробки, яку необхідно здійснити перш ніж медичний виріб можна буде використовувати (наприклад, стерилізація, остаточне складання);
10) стосовно медичних виробів, що випромінюють радіацію в медичних цілях, – докладна інформація про характер, тип, інтенсивність і поширення цього випромінювання.
Інструкції із застосування медичних виробів повинні містити відомості, що дають можливість медичному персоналу попереджати споживача про будь-які протипоказання та запобіжні заходи, зокрема інформацію про:
запобіжні заходи, що мають бути вжиті в разі зміни робочих характеристик медичного виробу;
запобіжні заходи, що мають бути вжиті у зв’язку з впливом магнітних полів, зовнішніх електричних впливів, електростатичних розрядів, тиском або змінами тиску, прискоренням, джерелами теплового займання тощо;
лікарський засіб чи засоби, для введення яких призначено відповідний медичний виріб, включаючи будь-які обмеження щодо вибору речовин для введення;
запобіжні заходи, що мають бути вжиті для усунення будь-яких спеціальних, незвичайних ризиків, пов’язаних з утилізацією медичних виробів;
лікарські засоби або похідні крові людини, які медичний виріб містить як невід’ємну частину;
ступінь точності, встановлену для медичних виробів з функцією вимірювання;
дату випуску або останнього перегляду інструкції із застосування.
Найпоширеніші символи які можуть зустрічатися на медичних виробах (вимоги та опис символів наведений в ДСТУ EN 980:2007 “Символи графічні для маркування медичних виробів”:
|
Символ |
Найменування символу |
Номер символу в ДСТУ EN 980:2007 |
 |
ОЗНАЙОМЛЕННЯ З ІНСТРУКЦІЯМИ ДЛЯ ЗАСТОСОВУВАННЯ |
5.8 |
 |
БІОЛОГІЧНІ РИЗИКИ |
5.9 |
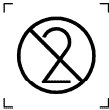 |
Повторно використовувати ЗАБОРОНЕНО |
4.2 |
 |
ЗАСТОРОГА! ОЗНАЙОМИТИСЯ ІЗ СУПРОВІДНИМИ ДОКУМЕНТАМИ Синонімом символу «Засторога! Ознайомитися із супровідними документами» є «Увага, дивись інструкцію з використовування» |
4.10 |
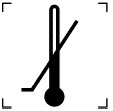 |
«НИЖНЯ МЕЖА ТЕМПЕРАТУРИ |
5.7.2 |
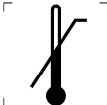 |
ВЕРХНЯ МЕЖА ТЕМПЕРАТУРИ |
5.7.1 |
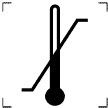 |
ТЕМПЕРАТУРНЕ ОБМЕЖЕННЯ |
5.7.3 |
 |
ВИКОРИСТАТИ ДО |
4.3 |
 |
ДАТА ВИГОТОВЛЕННЯ Цей символ повинен супроводжуватись датою, наведеною як зазначено в EN 28601, вона складається з чотирьох цифр року, двох цифр місяця і, за необхідності, двох цифр дня. Дата повинна бути написана поруч із символом. |
4.6 |
|
|
ВИРОБНИК Цей символ повинен супроводжуватися назвою та адресою виробника, який несе відповідальність за продукцію. Адреса не є необхідною за символу безпосередньо на контейнері, як визначено в EN 375 и EN 378, крім тих випадків, якщо контейнер безпосередньо також є зовнішнім контейнером. |
5.2 |
 |
КОД ПАРТІЇ Цей символ повинен супроводжуватися кодом партії виробника. Цей код буде суміжний із символом |
4.4 |
 |
НОМЕР ЗА КАТАЛОГОМ Номер виробника за каталогом має бути розташовано після або нижче символу, суміжного з ним. |
4.9 |
 |
РЕЄСТРАЦІЙНИЙ НОМЕР Цей символ повинен супроводжуватися реєстраційним номером виробника. Реєстраційний номер виробника повинен бути розташований після або нижче символу |
4.5 |
 |
СТЕРИЛЬНІСТЬ Цей символ — лише для медичних виробів, які повністю стерилізовані |
4.7 |
 |
Символ для стерильних медичних виробів, які оброблено із застосовуванням стерильної техніки |
4.11 |
 |
Символ методу стерилізації, під час якого використовують оксид етилену |
4.8.1 |
 |
Символ методу стерилізації, під час якого використовують випромінювання |
4.8.2 |
 |
Символ методу стерилізації, під час якого використовують пар чи сухий жар |
4.8.3 |
 |
МЕДИЧНИЙ ВИРІБ ДЛЯ ДІАГНОСТИКИ IN VITRO |
5.6 |
|
|
«МІСТИТЬ ДОСТАТНЬО ДЛЯ (n-) ВИПРОБОВУВАНЬ |
5.4 |


Медичні вироби для діагностики in vitro повинні маркуватись з дотриманням вимог пунктами 37-43 Розділу ІІ додатку І постанови КМУ від 2 жовтня 2013 р. № 754 «Про затвердження Технічного регламенту щодо медичних виробів для діагностики in vitro»:
Кожен виріб повинен супроводжуватися інформацією, необхідною для його безпечного і правильного використання з урахуванням підготовки та кваліфікації потенційних користувачів або споживачів, а також для ідентифікації його виробника.
Інформація, необхідна для безпечного і правильного використання виробу, розміщується безпосередньо на виробі та/або на упаковці для продажу. Якщо розміщення повної інформації на етикетці кожної одиниці виробу є практично неможливим, інформація повинна розміщуватися на упаковці та/або в інструкції для споживача/користувача, яка додається до одного або більше виробів.
Інструкція з використання повинна супроводжувати виріб або міститися в упаковці для одного або більше виробів.
Інструкція з використання виробу може бути відсутня, якщо виріб можна правильно та безпечно застосовувати без неї.
Інструкція з використання та етикетка складаються з урахуванням вимог Закону України “Про засади державної мовної політики”.
У разі можливості інформація, що повинна супроводжувати вироби, подається у формі символів. Усі використані символи та кольори повинні відповідати гармонізованим стандартам. Якщо відповідні стандарти відсутні, використані символи та кольори повинні бути описані у документації, що постачається разом з виробами.
Якщо вироби містять препарат, що може вважатися небезпечним з огляду на природу та кількість його складових, а також його форму, повинен бути нанесений символ, що позначає небезпеку, та здійснене маркування в установленому законодавством порядку. У разі відсутності достатнього місця для розміщення інформації на самому виробі або його етикетці відповідні символи небезпеки повинні розміщуватися на етикетці, а вся інша необхідна інформація – в інструкції із застосування.
Етикетка виробу повинна містити таку інформацію, що може наводитися у формі символів:
найменування або торгову марку та місцезнаходження виробника. Для виробів, що імпортуються з метою введення в обіг, на етикетці або зовнішньому пакуванні, або в інструкції із застосування додатково зазначається найменування та місцезнаходження уповноваженого представника, якщо виробник не є резидентом України;
інформацію, необхідну користувачеві або споживачеві для точної ідентифікації виробу та вмісту упаковки;
інформацію про особливий мікробіологічний стан або рівень чистоти (слово “Стерильно” або відповідний символ);
код партії виробів, зазначений після слова “Партія”, або серійний номер;
дату, до настання якої сам виріб чи його частина можуть безпечно використовуватися без погіршення експлуатаційних характеристик, у такому порядку: рік, місяць та в разі потреби – день;
до виробів для оцінки характеристик слова “тільки для оцінки характеристик”;
інформацію про призначення виробу для використання in vitro;
будь-які особливі умови зберігання та/або використання;
особливі інструкції із застосування;
відповідні попередження та/або заходи безпеки, яких слід вживати;
якщо виріб призначений для самоконтролю, цей факт повинен бути чітко зазначеним.
Якщо призначення виробу не є очевидним користувачеві або споживачеві, виробник чітко зазначає призначення такого виробу в інструкції із застосування та на його етикетці у разі наявності.
В обґрунтованих та практично можливих випадках вироби та їх окремі компоненти повинні ідентифікуватися, якщо необхідно – у рамках партії, з метою вжиття відповідних заходів для встановлення будь-якого потенційного ризику, який може бути спричинений такими виробами або їх компонентами.
Інструкції із застосування в разі доцільності містять:
інформацію, зазначену у пункті 40 цих Основних вимог, крім абзаців п’ятого і шостого пункту 40 цих Основних вимог;
якісний і кількісний склад реагентних виробів або дані про концентрацію активних інгредієнтів реагентів чи наборів реагентів, а також інформацію стосовно того, що виріб містить інші інгредієнти, які можуть вплинути на вимірювання;
умови зберігання та строк придатності після першого відкриття первинного пакування, а також умови зберігання та стабільність робочих реагентів;
експлуатаційні характеристики, зазначені у пункті 3 цих Основних вимог;
дані про необхідне спеціальне обладнання, в тому числі інформацію, необхідну для ідентифікації такого спеціального обладнання для належного застосування;
тип зразків, які повинні використовуватися для дослідження, будь-які спеціальні умови збирання, попередньої обробки та зберігання зразків, а також інструкції з підготовки пацієнта;
детальний опис процедури використання виробу;
процедуру вимірювання, необхідну для використання виробу, яка містить:
– принцип методу вимірювання;
– специфічні аналітичні характеристики виробу (в тому числі чутливість, специфічність, точність, повторюваність, відтворюваність, межі визначення та діапазон вимірювань, включаючи інформацію, необхідну для контролю відомих релевантних похибок), обмеження методу та інформацію про використання наявних референтних вимірювальних процедур та референтних матеріалів (стандартних зразків) користувачами;
– подробиці будь-яких подальших процедур або робіт, які необхідно проводити перед використанням виробів (наприклад, реконституції, інкубації, розведення, перевірки із застосуванням відповідних приладів тощо);
– інформацію про необхідність спеціальної підготовки користувача;
підхід, який застосовується для розрахунку аналітичних результатів;
інформацію про заходи, яких слід вжити у разі зміни аналітичних характеристик медичного виробу;
інформацію для користувача про внутрішній контроль якості, включаючи спеціальні процедури валідації та простежуваність калібрування виробу;
референтні інтервали для показників, що визначаються;
інформацію, необхідну для перевірки правильного встановлення виробу, правильного та безпечного його функціонування, а також додаткову інформацію про характер і періодичність технічного обслуговування та калібрування, необхідних для забезпечення правильної та безпечної роботи виробу; інформацію про безпечну утилізацію відходів;
інформацію про додаткові заходи або дії, які необхідно вчинити перед використанням виробу (стерилізацію, остаточне складання тощо);
необхідні інструкції на випадок пошкодження захисного пакування та інформацію про належні методи повторної стерилізації або знезараження;
інформацію про заходи безпеки, яких необхідно вживати в обґрунтовано передбачуваних умовах навколишнього середовища щодо впливу магнітних полів, зовнішніх електричних впливів, електростатичних розрядів, тиску або зміни тиску, прискорення, теплових джерел займання тощо;
інформацію про заходи безпеки, яких необхідно вжити для запобігання будь-яким специфічним, незвичайним ризикам, пов’язаним з використанням чи утилізацією виробів, включаючи спеціальні захисні заходи (якщо виріб містить речовини людського або тваринного походження, слід звертати увагу на їх потенційну інфекційну природу (інфектогенність);
дату випуску або дату останнього перегляду інструкції з використання;
специфікації для виробів для самоконтролю.
Якщо виріб призначений для багаторазового використання, інструкція із застосування повинна містити інформацію про належні заходи, які необхідно здійснити для повторного використання, включаючи промивку, дезінфекцію, пакування та повторну стерилізацію або знезараження, та про будь-які обмеження кількості повторного використання.
Якщо виріб повинен використовуватися в поєднанні з іншими медичними виробами або встановлюватися за допомогою медичних виробів чи під’єднуватися до них для функціонування за призначенням, інструкція із застосування повинна містити інформацію про характеристики таких виробів, достатню для ідентифікації відповідних медичних виробів або обладнання, які потрібні для отримання безпечного і правильного поєднання.
Результати роботи виробу повинні бути виражені та представлені так, щоб їх могли легко зрозуміти користувачі та/або споживачі без необхідної кваліфікації; інформація повинна містити поради користувачеві/споживачеві про те, що слід робити (у разі позитивного, негативного чи невизначеного результату), та вказівку на можливість хибного позитивного або хибного негативного результату.
Специфічні особливості можуть не зазначатися, якщо надана користувачеві/споживачеві інформація є достатньою для правильного застосування виробу для самоконтролю та розуміння ним отриманих результатів.
Зазначена інформація повинна включати чітку вказівку споживачу не приймати будь-які рішення медичного характеру (без попередньої консультації з лікарем).
Інформація повинна також містити вказівку на те, що, якщо виріб для самоконтролю використовується для моніторингу існуючого захворювання, пацієнт може застосувати лікування тільки після належної підготовки для здійснення такого лікування.
Активних медичні виробів, які імплантують повинні маркуватись з дотриманням вимог пунктами 12-16 додатку І постанови КМУ від 2 жовтня 2013р. № 755 «Про затвердження Технічного регламенту щодо активних медичних виробів, які імплантують
Вироби та їх комплектувальні частини повинні бути ідентифіковані для вжиття необхідних заходів після виявлення потенційного ризику у зв’язку з використанням виробів та їх комплектувальних частин.
Вироби повинні мати код, за яким вони та їх виробник можуть бути однозначно ідентифіковані (особливо щодо типу виробу та року виробництва). При цьому необхідно забезпечити можливість прочитати цей код без хірургічного втручання.
Якщо виріб або його комплектувальні частини містять вказівки, необхідні для забезпечення функціонування виробу, або визначають за допомогою візуальної системи параметри функціонування чи налаштування, така інформація повинна бути зрозуміла для споживача та користувача.
Кожен виріб повинен містити такі відомості (в разі можливості – у вигляді символів), нанесені чітко і розбірливо:
1) на стерильній упаковці:
метод стерилізації;
позначення, що дозволяє розпізнати упаковку;
найменування та місцезнаходження виробника;
опис виробу;
напис: “виключно для клінічних досліджень”, якщо виріб призначений для клінічних досліджень;
напис: “виріб, виготовлений на замовлення”, якщо виріб виготовлений на замовлення;
повідомлення, що виріб є стерильним;
місяць і рік виробництва;
зазначення строку придатності для безпечної імплантації виробу;
2) на зовнішній упаковці:
найменування та місцезнаходження виробника і найменування та місцезнаходження уповноваженого представника, якщо виробник не є резидентом України;
опис виробу;
призначення виробу;
характеристики для використання;
напис: “виключно для клінічних досліджень”, якщо виріб призначений для клінічних досліджень;
напис: “виріб, виготовлений на замовлення”, якщо виріб був виготовлений на замовлення;
повідомлення, що виріб є стерильним;
місяць і рік виробництва;
зазначення строку придатності для безпечної імплантації виробу;
умови транспортування і зберігання виробу;
для виробу, зазначеного у пункті 5 Технічного регламенту, зазначається, що виріб містить речовину, отриману з крові або плазми крові людини.
Під час введення в обіг кожен виріб має супроводжуватися інструкцією із застосування, в якій зазначаються:
рік надання дозволу на маркування знаком відповідності технічним регламентам;
інформація, зазначена в пункті 15 цього додатка (крім інформації щодо місяця і року виробництва та зазначення строку придатності для безпечної імплантації виробу);
параметри, передбачені виробником, та будь-які побічні ефекти від застосування виробу;
інформація, що дозволяє медичному працівнику обрати відповідний виріб, належне програмне забезпечення та комплектувальні частини;
інформація, що дозволяє медичному працівнику або споживачу застосовувати виріб, його комплектувальні частини та програмне забезпечення належним чином, а також інформація про характер, сферу застосування та час проведення контрольних перевірок і випробувань та/або про заходи з підтримання працездатності виробу;
інформація, що дає змогу уникнути окремих ризиків, пов’язаних з імплантацією виробу;
інформація про ризик взаємного впливу. Під ризиком взаємного впливу слід розуміти негативний вплив на виріб, спричинений приладами чи інструментами під час досліджень або лікування, і навпаки;
вказівки, необхідні у випадку пошкодження стерильної упаковки, та у разі можливості відомості про методи повторної стерилізації;
інформація про те, що виріб може бути застосований повторно (якщо це можливо) тільки у випадку, якщо він відновлений під відповідальність виробника до рівня відповідності встановленим вимогам.
Інструкція із застосування повинна також включати відомості, що дозволять медичному працівнику інформувати споживача про протипоказання та застережні заходи, яких слід вживати під час застосування виробу. Ці відомості повинні містити:
інформацію, що дає змогу встановити строк експлуатації джерела енергії;
застережні заходи, яких необхідно вжити у разі, коли спостерігатимуться зміни у функціонуванні виробу;
застережні заходи, яких необхідно вжити у випадку впливу дії магнітних полів, зовнішньої електростатичної індукції, електростатичних розрядів, збільшення або коливання тиску та прискорення в обґрунтовано передбачуваних умовах навколишнього середовища;
відповідну інформацію про лікарські засоби, для введення яких цей виріб розроблено;
дату видання або останньої редакції інструкції із застосування.
